
Tags
Tutorial Level
![]()
Absolute Quantification with EvaGreen®
The increasing emergence and dissemination of antibiotic resistance (AR) is a threat to human and animal health worldwide. Organic solid waste including animal manure and sewage sludge from wastewater treatment plants has been recognized as rich reservoir of both antibiotic resistant bacteria (ARB), and antibiotic resistant genes (ARG). The common practice of soil fertilization with animal manure or municipal waste contributes to the dissemination of ARB and ARG in the environment. There is a growing interest in ecological studies of antibacterial resistance owing to the increasing concern over the potential risks associated with community-acquired infections as well resistance gene transfer, not only among commensals but also to pathogens.
Tracking microbes and their gene content requires sensitive and reliable methods. One of the technical challenges is to combine sensitivity, specificity, and universality (all gene sequences have to be detected at once). Digital PCR (dPCR) using EvaGreen® offers such potential.
Here we choose to report the detection of the ermB gene, encoding for erythromycin resistance, in various organic wastes exposed to a cleaning process involving digestion and composting using dPCR.
This antibiotic is used on both human and animal foods. The ermB gene (364 bp) encodes an rRNA methylase that hinders antibiotic binding to the ribosome and usually results in cross-resistance to macrolides, lincosamides, and streptogramins B antibiotics. It is the most widely distributed gene among the erm gene class. It is present in a variety of Gram-positive bacteria, including enterococci, streptococci, and staphylococci. The potential for inter-species horizontal gene transfer (HGT) has facilitated the emergence of resistance in multiple species including Gram-negative bacteria of the genera Bacteroides, Shigella, Escherichia, Klebsiella, and Campylobacter.
Data presented in this tutorial has been obtained in collaboration with Dr. Nazaret Sylvie from UMR 5557 Ecologie Microbienne, CNRS, INRA, VetagroSup, UCBL, Université de Lyon, 43 Boulevard du 11 Novembre, F-69622 Villeurbanne, France.
1. How should I design this assay?
First of all, note that the rules for primers design are the same in qPCR and digital PCR.
During the PCR, primers select the region of interest to be amplified. EvaGreen®, a green fluorescent dye, binds to the amplified double strand DNA and becomes highly fluorescent.
For more details, please see the Primers & Probes and PCR Basic Knowledge items.
Designing Primers & Probes
Primer and probe design is a crucial step for a successful experiment.
The rules for designing primers and probes in a digital PCR assay are similar as for a qPCR assay:
1. Primers
Their length should be between 18 and 25 base pairs.
The criterion that you have to carefully monitor are:
- Percentage of GC: 40-60%
- Primer melting temperature (Tm): ideally between 55-65°C and Tm between both primers should not differ by more than 5°C
- G or C bases at the 3′ of the primer but not more than 2 in the last 5 bases
- Low probability of dimer or hairpin formation
2. Probes
The length should not exceed 30 base pairs. Ideally 15 base pairs for optimal specificity.
The criterion that you have to carefully monitor are:
- Percentage of GC: 20% to 80%
- Probe melting temperature: ideally from 4 to 10°C above the primer melting temperature
- Absence of more than 4 G repeats
- Low probability of dimer or hairpin formation
- Absence of G at the beginning of the probe
Sometimes, you will need to increase the Tm of the probe while keeping its size short. You can use modified bases such as locked nucleic acid (LNA) bases or peptide nucleic acid (PNA) bases. A probe with a minor groove binder (MGB) group is also an option to increase the Tm.
For further information about probe design, please refer to the following publications:
- Design of primers and probes for quantitative real-time PCR methods. Rodríguez A, Rodríguez M, Córdoba JJ, Andrade MJ, Methods Mol Biol. 2015;1275:31-56. doi: 10.1007/978-1-4939-2365-6_3. [PMID: 25697650]
- http://media.axon.es/pdf/78099_2.pdf
More information about MGB group, LNA, and PNA bases:
- Locked nucleic acids in PCR primers increase sensitivity and performance. Ballantyne KN, van Oorschot RA, Mitchell RJ. Genomics. 2008 Mar;91(3):301-5. doi: 10.1016/j.ygeno.2007.10.016.[PMID: 18164179]
- An introduction to peptide nucleic acid. Nielsen PE, Egholm M. Curr Issues Mol Biol. 1999;1(1-2):89-104. Review. [PMID: 11475704]
- https://academic.oup.com/nar/article/28/2/655/1039630
Tools for primer and probe designs:
PCR Basic Knowledge
“PCR” stands for Polymerase Chain Reaction.
PCR was first developed in 1983 by Kary Mullis, who was awarded the Nobel Prize in 1993 for this extraordinary invention. Indeed, PCR enables the clonal amplification of specific segments of DNA from a single fragment to thousand of millions of copies!
This tool is widely used in molecular biology for a variety of applications- manipulating DNA (cloning, mutagenesis), analyzing genes (though PCR-based sequencing for example), diagnosis or monitoring of pathologies, and also for parental testing or for DNA profiling in forensic studies.
1. What is in a PCR mix?
-
Polymerase
First and foremost, a PCR mix contains a thermostable polymerase, such as the ubiquitous Taq. The Taq polymerase was originally isolated from the thermophilic bacterium Thermus aquaticus in 1971 and is the original enzyme that was used for PCR by Kary Mullis. Since then, many other polymerases have been discovered. They have different properties regarding their tolerance to inhibitors, processivity (length of amplicon generated), speed of synthesis, etc. However, the Taq polymerase, whether in its original configuration or incorporating engineered elements, remains a favorite for PCR-based amplification.
-
dNTPs
The nucleotides are the building blocks of the reaction. They are present in the form of deoxyribonucleotides and are commonly referred to as dNTPs. The “dNTP” denomination means that dCTP (deoxycytidine triphosphate), dATP (deoxyadenosine triphosphate), dTTP (deoxythymidine triphosphate), and dGTP (deoxyguanosine triphosphate) are present in equimolar amounts. Depending on the application, the concentration of individual nucleotides may be further tuned, or some nucleotides may altogether be substituted with analogs, such as 5-fluorouracil or 6-mercaptopurine.
-
Reaction Buffer
Although the polymerase may be active over several pH points, the pH of most PCR commercial mixes is usually around 8 – 8.4 for optimal amplification. The pH of the reaction buffer is controlled by the use of a buffering agent, most commonly Tris-HCl. The reaction also comprises monovalent cations such as K+ and NH4+, divalent cations such as Mg2+, and less commonly Mn2+. The K+ salts neutralize the negative charge on the backbone of DNA and thus stabilize primer-template binding. Mg2+ is an essential co-factor of the polymerase. Its concentration in the reaction affects the specificity and efficiency of the reaction.
Other chemical species can be found in PCR mixes, such as detergents, additional salts, DMSO, or proteins such as bovine serum albumin. These compounds can be included to help with denaturation of the DNA strands, especially in sequences with high GC content, to improve polymerase activity, or to counter the inhibitory effects of substances present in the samples.
-
Primers
In order to amplify a DNA fragment by PCR, two DNA primers (single-stranded oligonucleotides) that are complementary to the 3’ ends of the sense and anti-sense strands of the DNA target are needed (Figure A). To know more about primers, refer to the “Designing Primers and Probes” item!
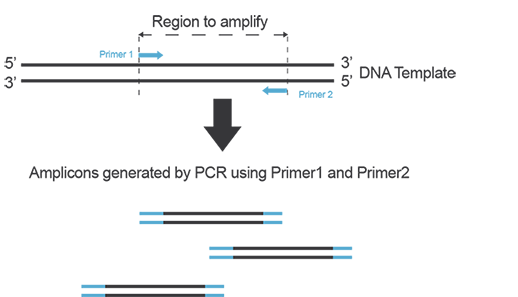
Figure A. Primers anneal to their target on each strand of DNA during PCR and yield amplicons defined by the hybridization location of each primer on the template.
-
Template DNA
Template DNA can be any DNA to be amplified, whether extracted from animal or plant cells, fungi, bacteria, or present in free form- for example circulating DNA in blood or urine. For optimal results, it is recommended to use long-enough purified DNA fragments for amplification to take place. Additionally, caution needs to be exercised to avoid chemical and/or UV-induced damage to the DNA template as it may affect your PCR results.
2. Thermal Cycling in PCR
PCR is based on thermal cycling: the reaction mix is subjected to sequential cycles of heating and cooling, which allow specific temperature-dependent reactions to occur.
-
Denaturation
First, DNA is denatured or melted at a high temperature, typically 94-96°C. During this step, the hydrogen bonds between complementary bases of the double-stranded DNA are broken, yielding two single-stranded molecules. Depending on the type of DNA, the reagents, and equipment used this step usually lasts ranging from 15 seconds to 1 minute and marks the start of each cycle of PCR.
-
Annealing
After denaturation of the template DNA strands, primers anneal to their target on the DNA template. The temperature of this step is critical to ensure specific hybridization of the primers to the target. In practice, this temperature is usually determined empirically- it needs to be low enough to allow primer binding but high enough for the binding to be specific to the intended target. Usually, annealing temperatures are between 3-5°C below the calculated Tm of primers, which translates to annealing temperatures for PCR between 50-65°C. Once the primer has annealed to the DNA, the polymerase binds to the primer-template hybrid and elongation can start.
-
Elongation
During elongation, the polymerase synthesizes new DNA strands by adding free nucleotides (dNTPs) present in the PCR reaction one-by-one, and the polymerization of the new strand complementary to the template takes place in the 5’ to 3’ direction
The temperature for elongation depends on the polymerase used, with 68-72°C being most commonly used. The length of the amplicon is taken into account when determining the time needed for the elongation step with a general rule of thumb of 1 minute per kb to be synthesized. The synthesis rate of the polymerase present in your PCR mix can be found in the manufacturer’s recommendation.
-
Additional steps
Initial denaturation
Before cycling starts, a longer initial denaturation step is often included (90-95°C for 1-10 minutes). Why? Because hot-start polymerases must be activated as these enzymes have been developed to reduce non-specific amplification early in the PCR reaction. Thus, they are locked in an inactive state by binding to an antibody or a chemical inhibitor. At elevated temperatures, a non-reversible dissociation occurs and this unlocks the enzyme.
Final Elongation
A final extension step (68-72°C for 5-10 minutes) can also be included to ensure all amplicons are fully elongated.
3. What happens during PCR?
-
Number of copies of target DNA fragment generated during PCR
If the reaction efficiency is 100%, the number of amplicons doubles after each cycle. This means that each copy of the DNA target initially present will be amplified 2n, where n is the number of cycles of PCR. Thus, when amplifying a single copy of your target region for 40 cycles, you could finally get 240 = 1.1 x 1012 amplicons!
-
Stages of PCR
Given that the number of amplicons doubles at each cycle when PCR efficiency is 100%, PCR amplification is referred to as exponential.
However, since this is an enzymatic reaction one or more factors will become limiting as the reaction progresses. Moreover, the polymerase may also start to lose activity. This will mean that at some point the amplification will start to level off, before reaching a plateau where no more products are generated.
4. How are the PCR results visualized?
-
Electrophoresis
Traditionally, the products of PCR are run on an agarose gel to visualize the amplicons. The agarose gel is stained with an agent that will bind to DNA (ethidium bromide or GelRed® for example) and illuminated with UV light in order to reveal the various DNA fragments present. Typically, a DNA ladder is run alongside the PCR products to estimate the size of the products, as well as the amount of DNA generated (Figure B).
Other electrophoresis techniques may use capillary gels and fluorescently labeled primers for fine discrimination of amplicon size.
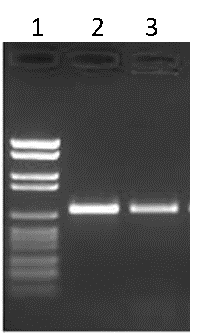
Figure B. Agarose gel electrophoresis. Lane 1: DNA ladder, Lane 2: PCR product 1, Lane 3: PCR product 2
-
Fluorescence
Different chemistries allow the fluorescence detection of DNA molecules. Amongst these chemistries, Hydrolysis probes, SYBR® Green I, and EvagGeen® are the most commonly used.
Hydrolysis probes (TaqMan® probes)
A probe, like primers, corresponds to a short oligonucleotide sequence (different from primer sequences) complementary to the target DNA sequence. Located between the sense and anti-sense primers, the hydrolysis probe consists of a fluorescent dye and a quencher. During amplification, the hydrolysis probe will be cleaved by the 3’->5’ exonuclease activity of the Taq polymerase. This will separate the fluorescent moiety from its quencher (Figure C). These fluorescent molecules will accumulate after each cycle. A fluorescent signal proportional to the number of amplicons bearing the probe hybridization sequence can be recorded. If you want to know more about probes, refer to the “Designing Primers and Probes” item.
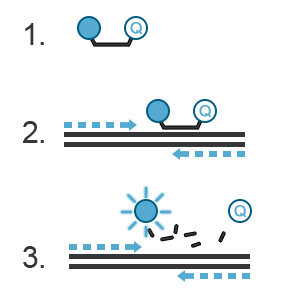
Figure C. Hydrolysis probes. 1. The probe is a single-stranded DNA sequence labeled with a fluorophore at its 5’ end and a quencher at its 3’ end. The fluorescence of the fluorophore is quenched, i.e., absorbed by the quencher- no light is emitted. 2. During PCR, the probe will hybridize with the amplicon generated in a sequence-specific manner. 3. The 3’ to 5’ exonuclease activity of the Taq polymerase destroys the hybridized DNA probe, thus freeing the fluorophore from the quencher. This fluorophore is now free to emit a fluorescent signal.
DNA-binding dye: EvaGreen®
EvaGreen® is a DNA binding dye that can be used to track PCR amplification in most digital PCR systems. It is a green fluorescent dye which becomes highly fluorescent when it is bound to double-stranded DNA. EvaGreen® is a thermoresistant molecule with excitation and emission spectra close to that of SYBR® Green I or 6-FAM dyes. Please note that the advantage of EvaGreen® over SYBR® Green I is its compatibility with droplet-based digital PCR systems. Thus, SYBR® Green I is mostly used in real-time PCR.
When using EvaGreen, the intensity of the fluorescent signal will correlate with the length of the amplicon generated. The longer the amplicon, the more bases available for EvaGreen binding, thus the stronger the fluorescence signal (Figure E).
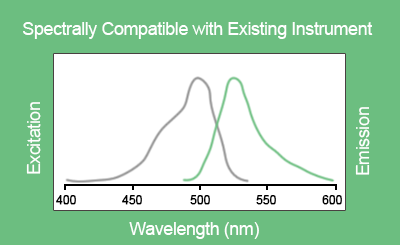
Figure D. Excitation and emission spectra of EvaGreen® in the presence of double-strand DNA in PBS buffer
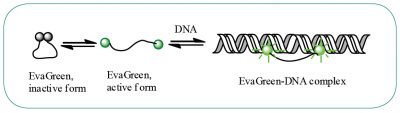
Figure E. EvaGreen® dye binds to double-strand DNA via a “release-on-demand” mechanism.
[https://biotium.com/technology/pcr-dna-amplification/evagreen-dye-for-qpcr/]
Fluorescence detection is the detection system used both in digital and real-time PCR.
Further information about EvaGreen® can be found on the manufacturer’s website
Digital PCR
Most digital PCR platforms use end-point PCR for quantification. End-point PCR means that the results of PCR are assessed when the plateau is reached. It occurs when the amplification has stopped due to loss of polymerase activity or an essential reagent (primers for example) present in the reaction.
In these reactions, fluorescent reporters are used. It can be DNA-binding dyes such as EvaGreen or fluorescently-labeled hydrolysis probes.
For further information on digital PCR refer to the “Principle of Digital PCR” item.
Real-time PCR
The intensity of the fluorescent signal is recorded at each cycle of PCR. The results are visualized on a graph where the intensity of fluorescence is plotted against the number of cycles. During the first few cycles, the fluorescent signal generated by the fluorescent reporter is indistinguishable from the background. As amplification takes place during cycles, the fluorescence eventually increases above noise level before tailing off and reaching a plateau.
Due to the accumulation of amplicons, the exponential increase of fluorescence can be visualized on the graph. A threshold for fluorescence intensity is placed at the beginning of this exponential phase. The cycle number at which the amplification curve intersects the threshold is called Cq (Cycle quantification). The quantification of an unknown sample assumes the use of a standard curve. By comparing the Cq obtained for DNA samples of known concentration with the Cq obtained for the unknown sample, DNA concentration in the unknown sample is then determined (Figure F)
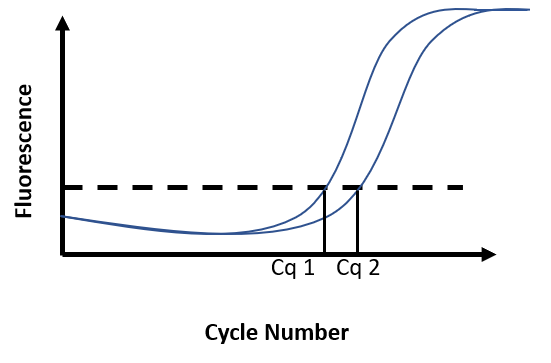
Figure F. Real-time PCR results. Fluorescence intensity is recorded at every cycle and plotted against the cycle number. Reaction 1 has a known DNA concentration (Cq1). At 100% efficiency, the DNA content in the reaction doubles at each cycle. Broadly, if Cq2 is distant from Cq1 from 1 cycle, this means there was half as much DNA in reaction 2 as there was in reaction 1 at the start of the PCR.
2. What do I need to perform this experiment?
For any digital PCR experiment, you need:
- A digital PCR system and dedicated consumables
- PCR mastermix containing the polymerase, dNTPs (desoxyribonucleotides), etc. (please check manufacturer’s recommendations)
- 1 reference dye if necessary (please check manufacturer’s recommendations)
- Purified water
- General lab supplies for PCR: microfuge tubes, vortex, microcentrifuge, etc.,
Then for this specific experiment, you will need:
- 1 primer set to amplify the ermB gene class
- extracted DNA from various organic wastes
- EvaGreen®, if not already included in your ready-to-use master mix (please check manufacturer’s recommendations)
Primer sequences we used for this assay can be found in Chen, J., Yu, Z., Michel Jr, F. C., Wittum, T., Morrison, M. (2007). Development and Application of Real-Time PCR Assays for Quantification of erm Genes Conferring Resistance to Macrolides-Lincosamides-Streptogramin B in Livestock Manure and Manure Management Systems. Applied and Environmental Microbiology, 73(14), 4407-4416. doi:10.1128/AEM.02799-06
3. How should I prepare my PCR mix?
Please note that this experiment has been done using the Naica™ System and the Sapphire chip.
a. Importance of DNA preparation
Once the DNA extraction has been done, you need to verify the DNA purity and quality of your samples.
For more details, please check the DNA preparation for Digital PCR item.
b. One more step before starting to manipulate: prepare your working sheet
If you are a qPCR user, the protocol for preparing the PCR mix is the same.
Here is what you need to know:
- how many samples do you want to run: for n=7 samples, in order to take into account the volume lost with pipetting error, prepare a total volume of PCR mix for n+1=8 samples.
- what are the manufacturer’s instructions for each component of the PCR mix?
Once you are ready, prepare your PCR mix as follows:
| Reagents | Final Concentration |
|---|---|
| PCR Mastermix (2X or 5X) | 1X or 0.75X, please refer to manufacturer’s instructions |
| Reference dye | Please refer to the manufacturer’s instructions |
| EvaGreen | Please refer to the manufacturer’s instructions |
| ermB primers | 200 nM |
| Organic Wastes DNA | unknown |
| Water | Up to 25 µL (depending on the manufacturer’s instructions) |
Table 1: PCR mix
In this experiment, we will quantify the number of ermB copies in 1 NTC and 6 samples:
Initial wastes A and B (wastes to be cleaned) are sewage sludge and pig slurry samples, respectively.
Intermediate wastes A-1 and A-2 were sampled after the anaerobic digestion (A-1) and the composting (A-2) treatments of the initial waste A.
Final wastes A and B (cleaned wastes) were obtained after anaerobic digestion and composting process.
Digital PCR DNA Preparation
DNA preparation is a crucial step for the success of your digital PCR assay. Template extraction, its quality control, and its storage are important parameters to monitor.
-
Template extraction
Digital PCR has been reported to be compatible with DNA & RNA templates extracted by various extraction methods used in laboratory- from phenol chloroform method to modern extraction kits1,2,3,4,5,6
-
Quality control
For optimal digital PCR performance, you should check the purity and the quality of your template. Using relevant methods such as spectrophotometry, you will be able to verify the good absorbance (A260/230 and A230/260) ratios of your extracted DNA solution. The presence of contaminants and/or potential inhibitors should be avoided.
-
Storage
The storage of your extracted DNA template is also an important factor to monitor. Indeed, for example, you need to avoid base degradation such as cytosine deamination and 8-Oxo-2′-deoxyguanosine formation due to oxidative damage since it may lead to base transversion during PCR amplification.
Buffering and temperature conditions have an impact on the quality of your sample. Storing at -20°C in Tris-EDTA is the standard condition, but please do not hesitate to take a look at the extraction kit manufacturer’s recommendations.
Other parameters, inherent to the sample itself, might have an impact on your digital PCR experiment and should be verified :
-
DNA with high GC content
If your template contains a GC-rich region, this might conduct to an incomplete amplification. Indeed, GC bonds are highly stable. Thus, in order to obtain a better denaturation of your template, you can try to add DMSO or betaine in your PCR mix.
-
High molecular weight DNA
High molecular weight DNA or plasmids have been reported by some manufacturers as problematic for partition generation. Thus, we recommend DNA shearing using chemicals or enzymatic methods prior to performing digital PCR. Please note that the digestion step might be performed directly in the PCR mix.
Finally, before starting your digital PCR assay you will have to proceed on the :
-
Conversion of mass of DNA in number of copies
If you are a qPCR user, you may be used to thinking in terms of mass of DNA input.
In digital PCR, you will need to think in terms of the number of copies of genes or genomes in your reaction volume.
In order to proceed on the conversion, the only thing you will have to know is the mass of the studied genome.
Then, simply apply this formula: Number of copies in reaction volume = mass of DNA in reaction volume (in ng)/ mass of the studied genome (in ng)
For more details about how to perform this calculation, refer to Part 3a of Rare Mutation Detection Tutorial.
Online calculators are also available :
- http://cels.uri.edu/gsc/cndna.html
- https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/dna-copy-number-calculator.html
Bibliography
1 Cai, Y., Li, X., Lv, R., Yang, J., Li, J., He, Y., & Pan, L. Quantitative Analysis of Pork and Chicken Products by Droplet Digital PCR. BioMed Research International, 2014, 810209. http://doi.org/10.1155/2014/810209. PMID: 25243184
2 Pérez-Barrios, C., Nieto-Alcolado, I., Torrente, M., Jiménez-Sánchez, C., Calvo, V., Gutierrez-Sanz, L., Palka, M., Donoso-Navarro, E., Provencio, M., Romero, A. Comparison of methods for circulating cell-free DNA isolation using blood from cancer patients: impact on biomarker testing. Transl Lung Cancer Res. 2016 Dec; 5(6):665-672. doi: 10.21037/tlcr.2016.12.03. PMID: 28149760
3 Demeke, T., Malabanan, J., Holigroski, M., Eng, M. Effect of Source of DNA on the Quantitative Analysis of Genetically Engineered Traits Using Digital PCR and Real-Time PCR. J AOAC Int. 2017 Mar 1;100(2):492-498. doi: 10.5740/jaoacint.16-0284. Epub 2016 Dec 22. PMID: 28118137
4 Holmberg, R.C., Gindlesperger, A., Stokes, T., Lopez, D., Hyman, L., Freed, M., Belgrader, P., Harvey, J., Li, Z. Akonni TruTip® and Qiagen® Methods for Extraction of Fetal Circulating DNA-Evaluation by Real-Time and Digital PCR. PLoS One. 2013 Aug 6;8(8):e73068. doi: 10.1371/journal.pone.0073068. Print 2013. PMID: 23936545.
5 Rajasekaran, N., Oh, M. R., Kim, S.-S., Kim, S. E., Kim, Y. D., Choi, H.-J., Byum, B., Shin, Y. K. Employing Digital Droplet PCR to Detect BRAF V600E Mutations in Formalin-fixed Paraffin-embedded Reference Standard Cell Lines. Journal of Visualized Experiments : JoVE, 2015, (104), 53190. Advance online publication. http://doi.org/10.3791/53190. PMID: 26484710.
6 Devonshire, A. S., Whale, A. S., Gutteridge, A., Jones, G., Cowen, S., Foy, C. A., & Huggett, J. F. Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Analytical and Bioanalytical Chemistry, 2014, 406(26), 6499–6512. http://doi.org/10.1007/s00216-014-7835-3. PMID: 24853859
c. Everything is ready: the hands-on part!
Prior to preparing your PCR mix make sure you work in a clean area in order to avoid all sources of DNA contamination. The use of a PCR hood is recommended. Assemble the PCR mix based on your working sheet. Make sure all the components are well homogenized before loading the PCR mix into the consumables used for partitioning.
Finally, the hands-on protocol will vary according to your digital PCR system. Please check the manufacturer’s instructions. If you are using the Naica™ System (Stilla Technologies) or the QX200™ Droplet Digital™ PCR System (Bio-Rad), you can check the “Performing Digital PCR Hands-On” item.
Performing Digital PCR Hands-On
Each digital PCR platform has its own hands-on protocol. In this item, we have chosen to describe protocols used on the Naica™ System and on the QX200™ Droplet Digital™ PCR System.
For both systems, we will assume that you have already prepared your PCR mix and divided it into 8 tubes: one per sample. You have also loaded your 8 samples.
1. Naica™ System:
Since the Sapphire chips are already prefilled with oil, you have only one last step before launching the PCR reaction.
To do so, you will need :
- 2 Sapphire Chips
- tips and pipette adapted for 25 µL
- 8 tall white caps (provided in the Sapphire Chips box)
Ready? Let’s go!
- Unpack the Sapphire Chips and remove the white caps
- Gently pipette your 25 µl of PCR mix containing your samples into each inlet port
- Finally, seal each inlet port with the tall white caps
The hands-on part is done and you are now ready to place your Sapphire Chips into the Naica™ Geode where the partition generation and the thermal cycling will occur.
A short movie of the workflow is also available here!
2. QX200™ Droplet Digital™ PCR System:
Here, we describe the manual protocol. A faster way to proceed would be to use the QX200™ AutoDG™ Droplet Digital™ PCR System where the following steps are automated and integrated.
Two more steps before launching the PCR reaction :
A. Partitions generation
To do so, you will need :
- 1 x DG8™ Cartridges for QX200™/ QX100™ Droplet Generator
- 1x DG8™ Cartridges holder for QX200™/ QX100™ Droplet Generator
- 1 x DG8™ Gasket for QX200™/ QX100™ Droplet Generator
- 1 x QX200™ droplet generator
- 1x Droplet Generation Oil for Probes
- Rainin pipettes and tips adapted for 20µL and 70 µL
Ready? Let’s go!
- Take the DG8™ Cartridges holder for QX200™/ QX100™ Droplet Generator. Open it by pressing the latches in the middle and sliding the two parts. Place one cartridge inside. Slide again in the original position to close
- Pipette your 20µL of PCR mix containing your sample in the middle wells of the cartridge. Then, pipette 70µL of droplet generation oil in the bottom wells
- Close the cartridge with one dedicated gasket
- Open the QX200™ Droplet generator by pressing the button on the top. Place the cartridge holder and close the droplet generator by pressing again the button
Partitions are now generated! The second and last hands-on part starts now :
B. Partitions transfer
To do so, you will need:
- 1 x ddPCR 96-Well Plate
- 1 x Pierceable foil plate seal
- 1 x PCR plate sealer
- Rainin pipettes and tips adapted for 40 µL
Let’s finish!
- Recover the DG8™ Cartridges holder from the Droplet Generator and remove the gasket. Do not open the cartridge holder.
- Gently pipette the 40µL of generated emulsion (top wells of the cartridge) and smoothly deliver it into a ddPCR 96-Well plate.
- Place the Pierceable foil on the ddPCR 96-well plate and proceed to the sealing part in the PCR plate sealer.
- Finally, place your ddPCR 96-well plate in the thermal cycler and launch your ddPCR program.
4. What steps are required to get my results?
a. Thermocycling
The ermB assay used in this example was optimized using the PerfeCTa qPCR ToughMix UNG mastermix on the Naica™ System (Stilla Technologies). In the table below you will find the thermocycling program used for the assay. If you are using a different mastermix or digital PCR system, you may have to optimize this protocol. For more details, please check “Digital PCR assay optimization” item.
| Cycles | Temperature | Time |
|---|---|---|
| 1 | 95 °C | 5 min |
| 5 | 94°C | 30 s |
| 63°C-59°C (Touchdown-1°C each cycle) | 30 s | |
| 72°C | 30 s | |
| 45 | 94°C | 30 s |
| 58°C | 30 s | |
| 72°C | 30 s |
Table 2: ermB assay cycling conditions. This program has been developed on a qPCR platform and directly transferred on the Naica™ System.
Touchdown PCR increases the specificity of the PCR reaction.
b. Data Acquisition
Depending on the system used, data acquisition will take the form of imaging a chip (Naica™ System, QuantStudio™ 3D Digital PCR System, CONSTELLATION® digital PCR system, etc.), or reading partitions one-by-one in a process analogous to flow cytometry (QX200™ Droplet Digital™ PCR System, Raindrop™ Digital PCR System). Please follow manufacturer’s instructions on how to perform this step.
Digital PCR Assay Optimization
Sub-optimal digital PCR settings very often lead to insufficient signal to noise ratio. This affects the separation between the negative and the positive partitions and prevents for an adequate threshold setting. That leads to limit the accuracy of the quantification and the sensitivity of the assay.
When developing an assay on a digital PCR system, there are several elements to consider:
- DNA template for assay optimization (positive control)
- Difference in fluorescence amplitude between negative and positive populations
- Presence of partitions of intermediate fluorescence (rain)
- Specificity – Are there non-specific populations? Can you detect positive partitions in your negative control?
1. DNA template for assay optimization
Ideally, the DNA template should be in the same form as it will be in the sample you will want to assay : sheared in small fragments if targeting circulating cell-free DNA or relatively intact if assaying genomic DNA. The DNA solution used should be devoid of contaminants and potential inhibitors and have good absorbance (A260/230 and A230/260) ratios. If there is no evident source for DNA material to develop your assay, you may use synthetic oligos as templates for optimization.
-
Presence of false positives
False positives are also a factor that contributes to the diminution of the sensitivity of the digital PCR assay. To limit this phenomenon, it is crucial to prevent DNA contamination in the laboratory or cross-contamination from well to well. Template quality is also a factor that is important to monitor as base degradation such as cytosine deamination and 8-Oxo-2′-deoxyguanosine formation due to oxidative damage may lead to base transversion during PCR amplification.
For more information about DNA preparation, have a look at the “DNA Preparation For Digital PCR” item.
2. Difference in fluorescence amplitude between negative and positive populations
When you run an assay for the first time, you might not distinguish the negative and positive populations on a digital PCR system (Figure A).
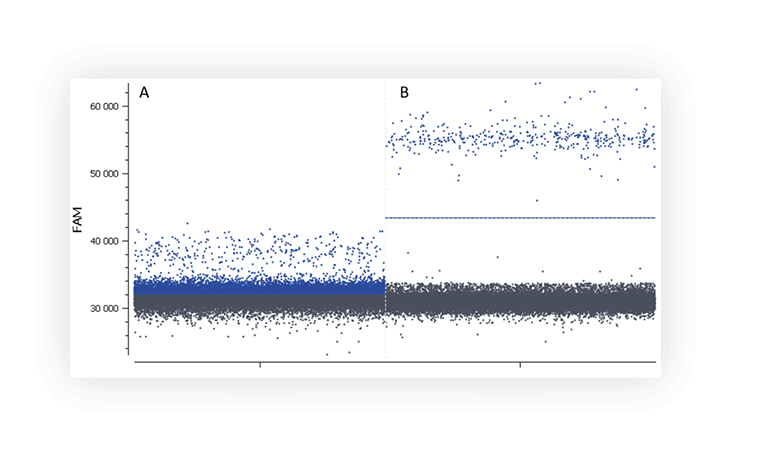
Figure A. The positive partition population (blue) is barely separated from the negative population (dark grey), rendering thresholding difficult on this 1D dot plot. B. The fluorescence of the positive population (blue) is clearly separated from the negative population (dark grey), placing a threshold is therefore unambiguous.
Here are a few tips to improve the separability of your populations:
- Check manufacturers recommendations on primers and probe concentrations to be used in your digital PCR system. These may be higher than the ones usually recommended in qPCR.
- Optimize hybridization temperature. Check for the highest hybridization temperature that gives you an optimal separability and no rain (see below).
- Check the type of probes you are using. Double-quenched probes will give you a lower basal fluorescence signal, and therefore a higher separability.
- Check whether your probes are too old! Depending on how long your probes have been stored, whether they have been stored properly, or whether they just encountered too many freeze-thaw cycles, it is possible that your probes are already hydrolyzed. In this case, you may see a high fluorescence basal level, and, since there are only a few intact probes for PCR, a low signal for the positive partitions.
- If using a system where you can vary acquisition parameters, you may also try to tune the exposure time
3. Presence of partitions of intermediate fluorescence (rain)
We call “rain” partitions that have intermediate fluorescence values between the positive and the negative populations. In these partitions, amplification/hybridization efficiency is sub-optimal. The rain makes the threshold setting harder, and thus may affect quantification.
Various factors may be at the origin of the “rain” such as, template degradation, PCR inhibitors, polymerase errors, primer dimer, target accessibility (Figure B) or non-homogenous distribution of fluorescence into partitions.
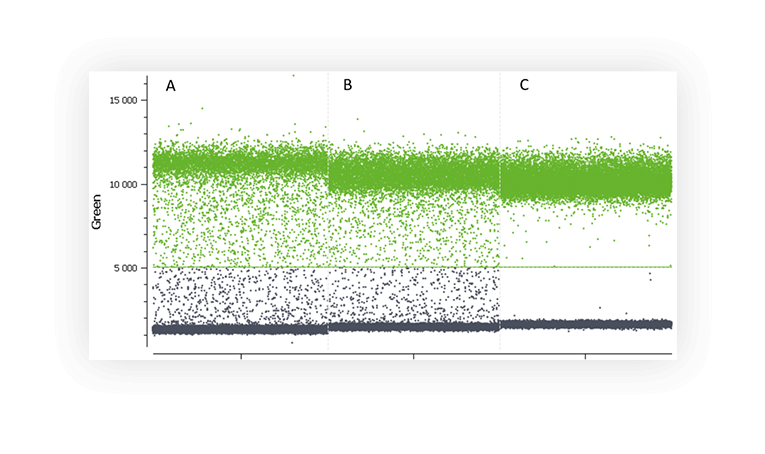
Figure B. 1D dot-plots of a digital PCR experiment in partition using one fluorescent assay targeting a non-digested plasmid (A and B) and a digested plasmid (C). Rain is clearly visible on the two first plot whereas plasmid digestion has removed most of the partitions corresponding to rain.
In order to minimize the rain, you can optimize your digital PCR reaction by:
- Checking the optimal hybridization temperature as described above
- Checking your DNA template for anything that may prevent accessibility to the target. Is this a GC-rich region? Try additives such as DMSO or betaine. When using high molecular-weight DNA or plasmids, it is also best to fragment the DNA by digestion or mechanical means prior to digital PCR. If using digestion, you may also be able to perform this step directly in the mix.
- Making sure to use DNA free from inhibitors
- Increasing the number of cycles in order to ensure that all partitions reach the reaction plateau.
4. Presence of non-expected populations
When assaying a single target, you usually expect a single positive population. But sometimes, you will discover that a second distinct population has appeared (Figure C). It can be due to primer dimers and non-specific amplification.
Multiple fluorescence populations may prevent from setting a proper threshold.
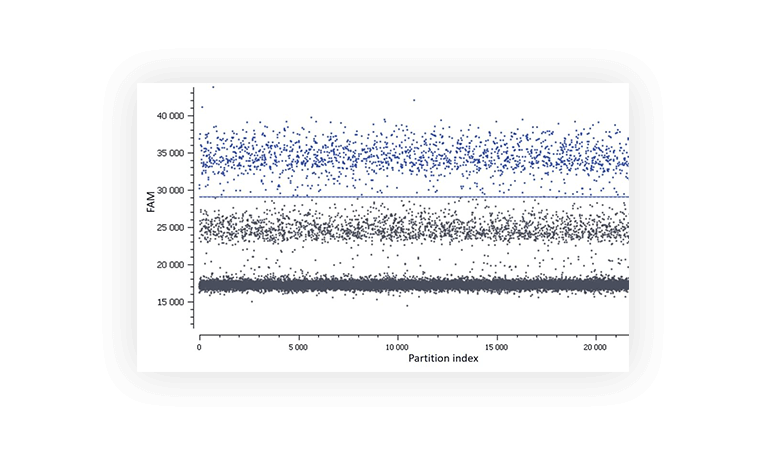
Figure C. In this assay, a second population is observed between the negative (lower grey band) and the positive (blue).
Various methods can be employed to limit this phenomenon such as optimization of primer design, increase of annealing temperature and touchdown PCR.
This specificity issue may be fixed:
- The hybridization temperature is potentially too low so make sure you pick the highest temperature that gives you optimal separability while minimizing the rain
- Have you thought about performing a touchdown PCR?
- Is this non-specific population really an issue ? In terms of quantification, you can just decide to place the threshold above it as we have done in Figure C. The non-specific population amplified will just be considered part of the negative population. The results will thus be calculated only for the amplified partitions that show a reaction efficiency consistent with a specific hybridization of the primers and probes. Please note that this is not the optimal way of analyzing your data since it can have an impact on precision and sensitivity.
- Re-design your primers. To do so, you may use online tools to check that your primers and probes do not hybridize elsewhere in the genome.
For more information on optimization to improve your digital PCR assay, see the following article:
Lievens, A., Jacchia, S., Kagkli, D., Savini, C., Querci, M. Measuring Digital PCR Quality: Performance Parameters and Their Optimization. PLoS One. 2016 May 5;11(5):e0153317. doi: 10.1371/journal.pone.0153317. eCollection 2016. PMID: 271494
5. How should I analyze my data?
In an EvaGreen digital PCR experiment, you have only one fluorescence channel to check so your data will be represented as a 1D dot-plot (Figure A).
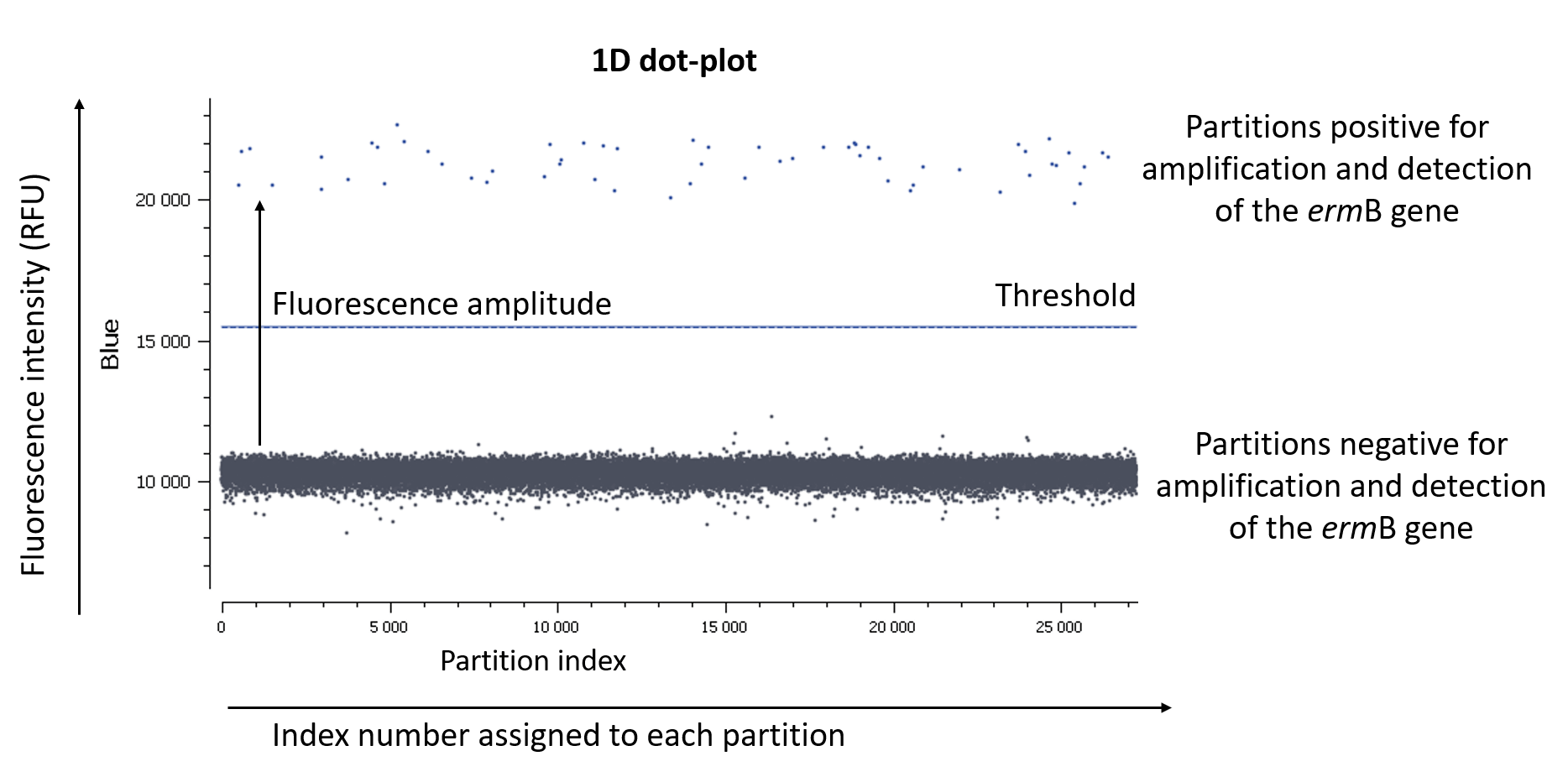
Figure A: The usual visualization of data in EvaGreen digital PCR experiments is by using a 1D dot-plot, in which each dot represents one partition. The y-axis gives the fluorescence intensity of each partition in the blue detection channel, while the x-axis gives the index number assigned to each partition by the analysis software.
For more details, please check the Fluorescence considerations with Evagreen® dye item.
a. Quality controls
Depending on the type of data acquisition, data analysis software can present different quality controls. Two criteria should be carefully considered:
- Check the NTC: none or very few positive partitions are accepted. As you can see in Figure D, our NTC presents only negative partitions.
- Check the total number of analyzed partitions: a high number of analyzable partitions will allow you to decrease the uncertainty associated with the measured concentration. In this assay, as you can see in table 3, we finally obtained between 25998 and 28269 partitions allowing us to validate our experiment.
On the Naica™ System, you can also visualize the generated partitions within the well. This will allow you to:
- Verify that the positive partitions are randomly distributed and thus, follow a Poisson distribution
- Verify the appearance of the partitions (size, level of fluorescence,etc.)

Figure B: Using the analysis software included in the Naica System, you can visualize the generated partitions for each sample. These partitions are self-assembled in a 2D monolayer array. The well is here called “chamber” and the pillars maintain its structure. The area in the top left corner appears empty: in order to avoid stacking, partitions do not completely fill the chamber. Three colors are used to distinguish artifacts (red crosses) from negative (green crosses) and positive (green crosses surrounded by blue circles) partitions. Artifacts are automatically excluded from the analysis. It can correspond to impurities like dust on the surface of the chamber (1). Partitions distributed around pillars (2) and the chamber (3) might be affected by the “edge effect” so the software identifies them as artifacts. Finally, isolated partitions (4) are also excluded. The white square represents the area presented in Figure B.

Figure C: Here, we zoomed in the area represented by a white square in Figure A. The legend can be removed so partitions can be easily visualized.
b. Threshold setting
Usually, the threshold is automatically set by the analysis software using a clustering algorithm that maximizes (resp. minimizes) inter-cluster (intra-cluster) point-to-point distances. Partitions above the threshold are positive and those under the threshold are negative. Thus, setting thresholds is a mandatory step to process to the quantification results by digital PCR.
Since the threshold setting is an automated calculation, we advise you to have a look at the dot-plots.
Indeed:
- when there is a very small number of positive partitions or a very small number of negative partitions, automated thresholding tends to fail out. Thus, the threshold has to be manually set
- when you observe a lot of partitions between the positive and the negatives ones (a phenomenon called “rain”), you might have to adjust the threshold as well (please note that you should also consider optimizing your assay if the rain is really important)
In this experiment, the threshold has been correctly set by the analysis software (Figure D).
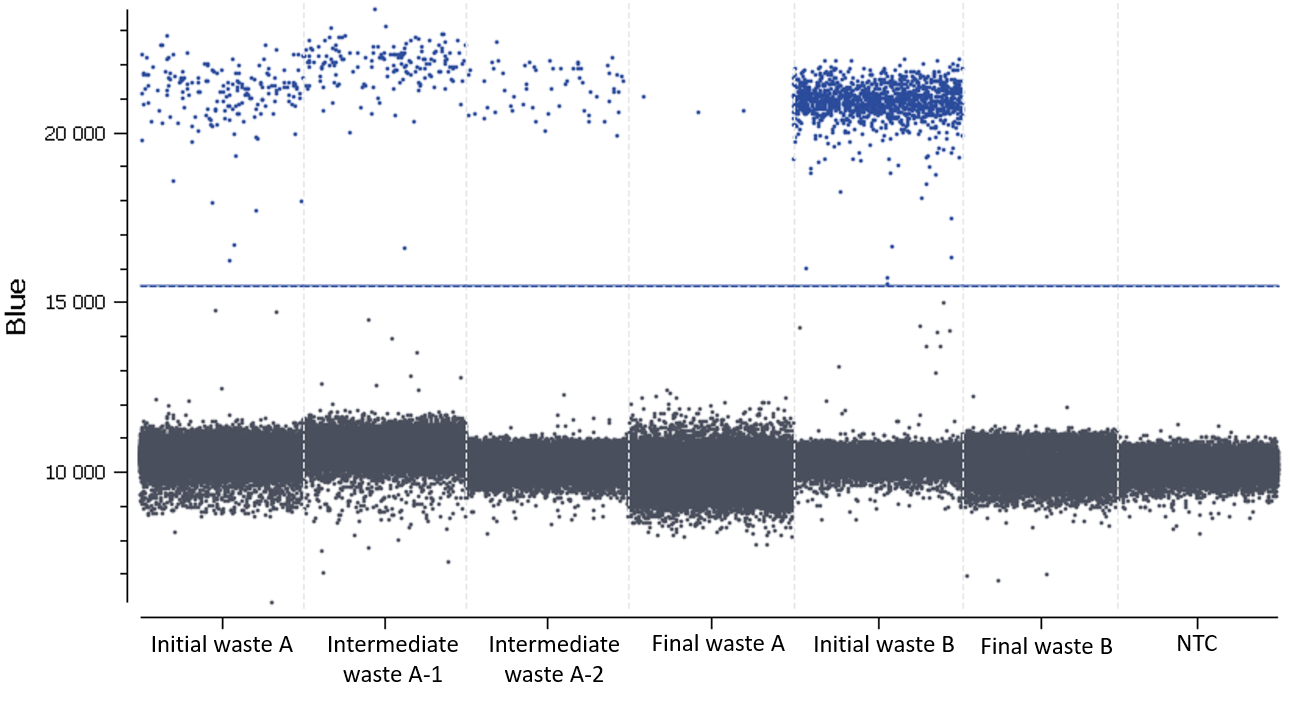
Figure D: 1D dot-plots for all tested samples and the NTC. The threshold is represented by a blue line. The negative and positive partitions are clearly separated so the analysis software was able to correctly set the automated threshold.
Fluorescence considerations with EvaGreen® dye
Fluorescence intensity of the positive signal
The fluorescence intensity of positive partitions in digital PCR experiments with EvaGreen® depends on the length of the double-stranded DNA amplicons that are generated during PCR. At equivalent PCR efficiencies and primer concentrations, long amplicons will produce a more intense fluorescent signal than short ones (Figure A). This is explained by the fact that more Evagreen® molecules will bind to the long amplicons relative to the short amplicons.
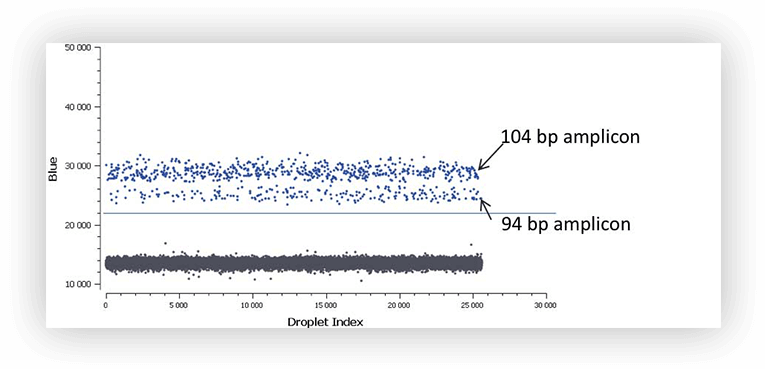
Figure A. At equivalent PCR efficiencies and primer concentrations, the 104 bp amplicon will produce a more intense fluorescent signal than the 94 bp amplicon.
Fluorescence background
Different reasons can explain a higher fluorescence background:
- Presence of single-strand DNA: what you must keep in mind is that although EvaGreen® dye is highly fluorescent when it is bound to double-stranded DNA, a fluorescence background can be observed in presence of single-stranded DNA which can be caused by primer dimers. To minimize this effect, make sure you design short primer sequences that will not form dimers, and that the final concentration of your primers in the PCR mix does not exceed 250 nM.
- The high molecular weight of your DNA sample: if you are working with human DNA for example, the EvaGreen® dye will bind to the starting material. Even if this starting material is present in low concentrations, it will be capable of capturing large amounts of EvaGreen® per DNA molecule. In turn, this may prevent adequate separability of negative and positive populations after PCR amplification. To limit this phenomenon, the initial input of DNA in your digital PCR experiments should be assessed with respect to the fluorescent signal generated by the positive partitions and you should avoid a high concentration of DNA.
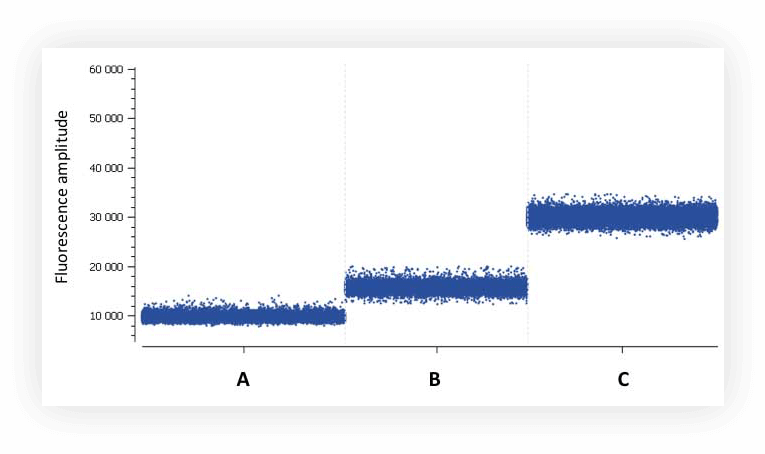
Figure B. Background fluorescence of partitions in digital PCR using EvaGreen® chemistry. A: master mix with no DNA template and no primers, B: master mix with 15 ng human DNA template and no primers, C: master mix with 200 nM primers and no DNA template.
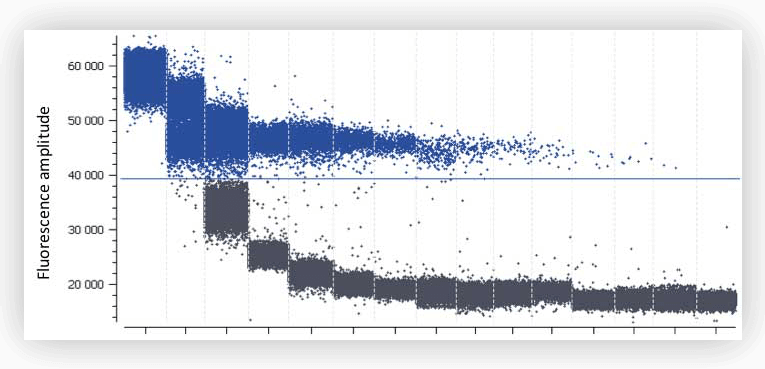
Figure C. Digital PCR experiment using EvaGreen® on a serial dilution of human genomic DNA. High background fluorescence can be observed with the 3 first dilutions containing 130, 66 and 33 ng of DNA respectively.
c. Calculating target concentration
The threshold being set, the software automatically quantifies the number of positive and negative partitions for each target. This digital readout is then converted to a result in the number of copies of the target per microliter using the Poisson distribution.
Each result is provided together with a 95% confidence interval which reflects the precision of the measurement. The accuracy of the measure can also be given by the “95% Confidence interval” value. The smaller, the better!
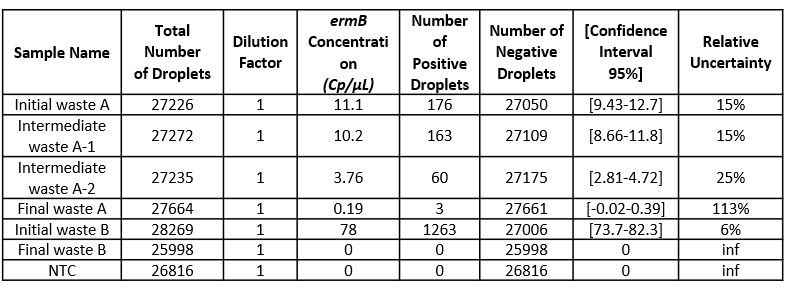
Table 3: For each sample concentration its 95% interval of confidence and its associated relative uncertainty is automatically calculated by the analysis software. Here, the dilution factor is 1 so the data obtained is the concentration in the well. Please note that if you prefer to obtain the concentration in your stock solution, do not forget to correct this value by your dilution factor.
In conclusion, data showed that both sewage sludge and pig slurry i.e. Initial wastes A and B, respectively, harbor ermB genes and that the cleaning processes led to the complete or almost complete elimination bacteria carrying this gene.
6. To sum up…
- Simply assay your samples with the conditions set up above, or optimized conditions if using another digital PCR system.
- Make sure you follow the MIQE guidelines if you are designing your own assay (For more details, please see the “MIQE Guidelines” item.)
- Verify that the threshold is placed adequately on your dot-plots.
- Export your results from the manufacturer’s software, making sure to include for each sample: the partition volume, the total number of partitions, as well as the number of positive partitions.
Congratulations for successfully following this digital PCR experiment!
Want to discover more dPCR experiments? Check our other tutorials!
MIQE Guidelines
In order to ensure experimental transparency, consistency between research laboratories and thus, maintain a high level of integrity in publications, a guideline for real-time PCR experiments has been edited in 2009 by an international research team [PMID: 19246619].
Considering the increasing number of publications in the Digital PCR field, the MIQE (for Minimum Information for Publication of Quantitative real-time PCR Experiments) guideline has been updated for digital PCR experiments under the name “the Minimum Information for the Publications of Quantitative Digital PCR Experiments guidelines (dMIQE)”[PMID: 23570709].
Having a look at this guideline before starting designing an experiment can be very informative. Indeed, as an example of what you can read in the dMIQE, you will find, below (Table 1), a checklist you might follow for performing a digital PCR experiment. Finally, all items are categorized as essential (E) or desirable (D) for manuscript submission. Thus, citing the following dMIQE in your publication will be a guarantee of quality, for reviewers and readers.
| Table 1. dMIQE checklist for authors, reviewers and editors.a | |||
| Item to check | Importance | Item to check 2 | Importance |
| Experimental design | dPCR oligonucleotides | ||
| Definition of experimental and control groups. | E | Primer sequences and/or amplicon context sequence.b | E |
| Number within each group. | E | RTPrimerDB (real-time PCR primer and probe database) identification number. | D |
| Assay carried out by core lab or investigator’s lab ? | D | Probe sequences.b | D |
| Power analysis. | D | Location and identify of any modifications. | E |
| Sample | Manufacturer of oligonucleotides. | D | |
| Description. | E | Purification method. | D |
| Volume or mass of sample processed. | E | dPCR protocol | |
| Microdissection or microdissection. | E | Complete reaction conditions. | E |
| Processing procedure. | E | Reaction volume and amount of RNA/cDNA/DNA | E |
| If frozen-how and how quickly? | E | Primer, (probe), Mg ++ and dNTP concentrations. | E |
| If fixed- with what, how quickly? | E | Polymerase identity and concentration. | E |
| Sample storage conditions and duration (especially for formalin-fixed, paraffin-embedded samples). | E | Buffer/kit catalogue no. and manufacturer. | E |
| Nucleic acid extraction | Exact chemical constitution of the buffer | D | |
| Quantification-instrument/method. | E | Additives (SYBR green I, DMSO, etc.). | E |
| Storage conditions: temperature, concentration, duration, buffer. | E | Plates/tubes Catalogue No and manufacturer. | D |
| DNA or RNA quantification | E | Complete thermocycling parameters. | E |
| Quality/integrity, instrument/method, e.g. RNA integrity/R quality index and trace or 3’:5’. | E | Reaction setup. | D |
| Template structural information. | E | Gravimetric or volumetric dilutions (manual/robotic). | D |
| Template modification (digestion, sonication, preamplification, etc.) | E | Total PCR reaction volume prepared. | D |
| Template treatment (initial heating or chemical denaturation). | E | Partition number. | E |
| Inhibition dilution or spike. | E | Individual partition volume. | E |
| DNA contamination assessment of RNA sample. | E | Total volume of the partitions measured (effective reaction size). | E |
| Detail of DNase treatment where performed. | E | Partition volume variance/SD. | D |
| Manufacturer of reagents used and catalogue number | D | Comprehensive details and appropriate use of controls. | E |
| Storage of nucleic acid: temperature, concentration, duration, buffer. | E | Manufacturer of dPCR instrument. | E |
| RT (if necessary) | dPCR validation | ||
| cDNA priming method + concentration. | E | Optimization data for the assay. | D |
| One- or 2-step protocol. | E | Specificity (when measuring rare mutations, pathogen sequences etc.) | E |
| Amount of RNA used per reaction | E | Limit of detection of calibration control. | D |
| Detailed reaction components and conditions. | E | If multiplexing, comparison with singleplex assays. | E |
| RT efficiency. | D | Data analysis | |
| Estimated copied measured with and without addition of RT.b | D | Mean copies per partition (ʎ or equivalent). | E |
| Manufacturer of reagents used and catalogue number. | D | dPCR analysis program (source, version). | E |
| Reaction volume (for 2-step RT reaction). | D | Outlier identification and disposition. | E |
| Storage of cDNA : temperature, concentration, duration, buffer. | D | Results of no-template controls. | E |
| dPCR target information | Examples of positive(s) and negative experimental results as supplemental data. | E | |
| Sequence accession number. | E | Where appropriate, justification of number and choice of reference genes. | E |
| Amplicon length. | E | Number and concordance of biological replicates. | D |
| In silico specificity screen (BLAST, etc.) | E | Number and stage (RT or dPCR) of technical replicates. | E |
| Pseudogenes, retropseudogenes or other homologs? | D | Repeatability (intraassay variation). | D |
| Sequence alignment. | D | Reproductibility (interassay/user/lab etc. variation). | D |
| Secondary structure analysis of amplicon and GC content. | D | Experimental variance or Cl.d | E |
| Location of each primer by exon or intron (if applicable). | E | Statistical methods used for analysis. | E |
| Where appropriate, which splice variants are targeted? | E | Data submission using RDML (Real-time PCR Data Markup Language).
|
D |
| a All essential information (E) must be submitted with the manuscript. Desirable information (D) should be submitted if possible.
b Disclosure of the primer and probe sequence is highly desirable and strongly encouraged. However, since not all commercial predesigned assay vendors provide this information, when it is not available assay context sequences must be submitted [Bustin et al. (48)]. c Assessing the absence of DNA using a no-RT assay (or where RT has been inactivated) is essential when first extracting RNA. Once the sample has been validated as DNA free, inclusion of a no-RT control is desirable, but no longer essential. d When single dPCR experiments are performed, the variation due to counting error alone should be calculated from the binomial (or suitable equivalent) distribution. |
|||